Model-Informed Drug Discovery and Development in Clinical Development
As a therapeutic progresses toward the clinic, there are many questions that need to be solidified as a project team prepares for first-in-human studies. The answer to many of these questions is unknown and modeling and simulation helps predict them.
Human Dose Predictions
While dose predictions can be, and some would argue should be, estimated throughout R&D, confidence in these predictions grows as you progress through the pipeline as additional data is generated and integrated into the mechanistic model. Various factors can impact dose such as target properties, indication properties, drug properties, and acceptable toxicities or safety. In the absence of human data, modeling and simulation represents a holistic approach of integrating available knowledge, preclinical data, and competitor or comparator clinical data for the purpose of providing superior human dose predictions.
When predicting a first-in-human (FIH) dose, preclinical efficacy and toxicology data help shape the therapeutic window. Efficacy is achieved when a drug elicits a desired response and safety (non-toxic) is the range under which you do not elicit any undesired adverse events. For biologics, when considering efficacy, many groups will calculate and report their minimum anticipated biological effect level, or MABEL, as the minimum dose needed to start seeing some type of pharmacodynamic effect. When considering safety, it’s important to understand at what dose level you might start to see side effects, whether that’s cytokine release, apoptosis, neutropenia, etc. Many groups will report a NOAEL, or no observable adverse effect level. The MABEL and NOAEL calculations are based on preclinical in vitro or animal in vivo studies and must be translated to a dose appropriate for humans.
Species Translation
Traditionally allometric scaling is used to scale preclinical dose calculations to a human dose using a factor such as body size. However, this approach is a very one-size-fits-all approach and cannot compensate for the variability often seen in large patient populations or with complex therapeutics, or are not known for the most novel and complex therapies (e.g., cell and gene therapies). Protein therapeutics offer very potent approaches to treating certain indications, but they also have complex relationships between parameters such as binding potency, exposure, and efficacy dosing on the timescales of biological responses. These complex relationships bring challenges scaling from preclinical data to predicting efficacious and safe doses, and selecting dosing regimens for clinical studies.
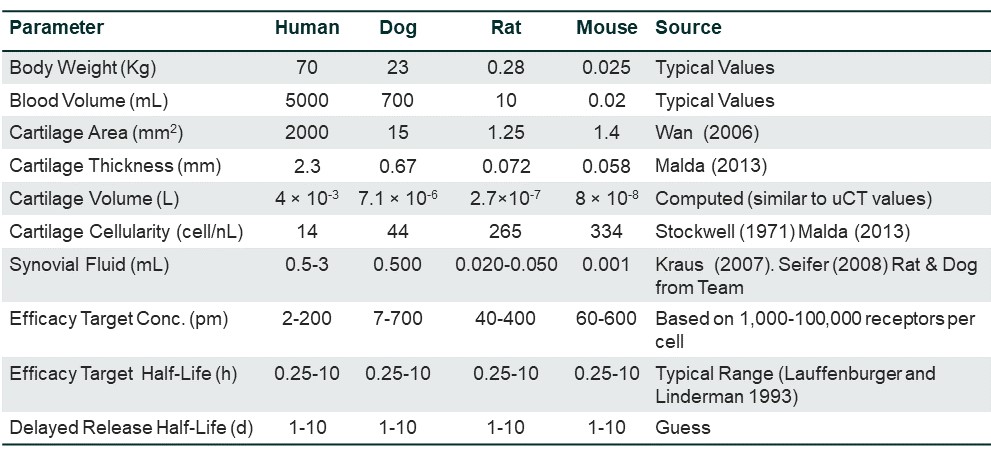
Parameters can vary significantly across species, within indications, and across indications in humans, and the impact of this change is not always obvious or straightforward. In one example, where a model was developed for Osteoarthritis, understanding the differences in parameters between species became key. The figure below shows how a mouse has a much smaller cartilage area compared to humans, but the cartilage cellularity is much higher in mice than the other species. If you were going to predict dose from mice, then you would be under-dosing humans due to the lack of cartilage cellularity. Depending on the disease area, patients may be losing cartilage. It’s important to do this work early on to get accurate dose predictions for efficacy in clinical trials.
As described previously, mechanistic modeling includes biophysical parameters related to the therapeutic and the disease system such as binding affinity, drug half-life, receptors per cell, etc. This makes it possible to better scale from in vitro or preclinical in vivo data and experiment using real, biophysical parameter human values to increase the accuracy of the resulting dose prediction.
Project Optimus
The FDA’s Oncology Center of Excellence (OCE) announced an initiative titled Project Optimus aiming to reform the dose optimization and selection process in oncology to be more appropriate for target therapies, such as biotherapeutics. This is so important, as for many patients using investigational therapies, this is often the last effort to save a patient’s life and starting at homeopathic doses may not help the patients, but the doses must be safe. Modeling and simulation approaches are beneficial to Project Optimus by allowing stakeholders to take a step back and think about various what-if questions such as:
- What if the target is under or overexpressed tenfold?
- What if you have target mediated drug disposition (TMDD)? How would that impact target coverage mechanistically?
- How can I predict more accurate therapeutic index (TI) and efficacy thresholds?
In thinking about the questions above, it would also be beneficial to build a mechanistic population PK (Mechanistic PopPK) model or simulated patient variability using mechanistic PK/PD modeling. This provides understanding of uncertainty and variability in patient parameters coupled with the therapeutic parameters and how that would impact predictions on safety and efficacy. At times this could allow justification of higher safe starting doses in comparison to starting doses that could be found using traditional methods, such as the minimum anticipated biological effect level (MABEL) and the no observed adverse effect level (NOAEL).
Pre-Investigational New Drug (IND) and IND Guidance
MID3 can help address Pre-IND and IND questions, such as:
- How does my program compare against the standard of care or future standards of care?
- How do I select the starting dose for my first-in-human study?
- If starting in Healthy Volunteers, what is the predicted efficacious dose for my molecule, gene therapy, or cell therapy?
- What is my SAD and MAD strategy?
- How do I translate from Healthy Volunteers into patient Populations? Across indications?
- What is R2PD dose based on phase 1 data?
- How do we optimize dosing to maximize the therapeutic window?
- What patients? Indications?
- Why didn't my trial work? What does our backup candidate look like?
- How do we differentiate our candidate from competitors?
MID3 in the Clinic
Once in the clinic, it is common to have clinical data coming in but the patient numbers might be small so it’s typically not enough data to understand efficacy from an empirical, statistical and population point of view. Mechanistic modeling and simulation approaches allow you to answer more questions earlier where you typically would have to wait for more patients, typically in later clinical trials.
MID3 can help design and support clinical trials by, for example, determining patient selection, sample collection times, biomarker selection, and how high and what frequency of a dose is required, or number of cells to deliver to assess proof of clinical concept and proof of clinical mechanism. Clinical trial simulations can be used to compare the characteristics of different designs, and maximize the probability of success for a trial. A model-based approach to sample size determination offers advantages over traditional power-based analyses by accounting for differences in patient characteristics between trials, disease progression, and nonlinear exposure-response relationships. The probability of false positive or false negative results associated with particular clinical trial designs can also be probed through simulation of, for example, population PK/PD (PopPK/PD), mechanistic PopPK/PD and Virtual Populations, PK/PD, and ER models, and the risks of these outcomes can be minimized. The impact of increasing or decreasing the dose or frequency of administration and route of administration can be assessed. Doses, regimens, and sampling times can be chosen such that the information gleaned from the trial is maximized, facilitating better future trials.
Example of MID3 in Clinical Development
Challenge: A team was developing a novel therapy to treat a rare disease, Crigler-Najjar syndrome type 1 (CN1), caused by a marked decrease in uridine-diphosphateglucuronosyltransferase (UGT1A1) enzyme activity. The absence of UGT1A1 causes hydrophobic unconjugated bilirubin to clear slowly, which causes its accumulation in the circulation, deposition into tissues, and transport through the blood-brain barrier (BBB). Accumulation in the brain puts patients at high risk for neurological deficits. The novel therapy would deliver a modified messenger RNA encoding of UGT1A1 as a lipid nanoparticle (LNP) to restore hepatic expression of UGT1A1, allowing normal glucuronidation and clearance of bilirubin in CN1 patients. The mechanism of action of this modality and clinical pharmacology are very complex (multi-step kinetic processes in different time scales in different tissues). Because of the complexity, the team needed to take a model-based approach to capture all of the biological and pharmacological knowledge in a quantitative framework to support the translation from preclinical to clinical studies, and first-in-human studies.
How MID3 Helped: In this project, MID3 helped justify the elimination of some biomarkers as non-informative and not required for establishing PK/PD relationships, informed the recommended clinical dose range and safe starting dose, impacted the clinical strategy in terms of patient populations for Phase 1, and projected consequences of nonlinear parameters that do not follow typical allometric scaling rules to inform first-in-human dose.